

 中 文
中 文

Tackling the Talent Crunch: Strategies for Staffing Shortages in Practices
2025-09-27
2026-07-06
If you manufacture, distribute, or purchase medical devices in Europe, you've likely encountered the terms MDD and MDR. Although both relate to CE marking for medical devices, they represent two different regulatory frameworks with significantly different requirements.
Since the Medical Device Regulation (EU) 2017/745 (MDR) became fully applicable on 26 May 2021, it has gradually replaced the former Medical Device Directive (93/42/EEC, MDD). The transition has introduced stricter requirements for clinical evidence, product traceability, post-market surveillance, and technical documentation.
For manufacturers of dental CAD/CAM materials and equipment, understanding these differences is essential for maintaining compliance and accessing the European market.
The Medical Device Directive (93/42/EEC) was introduced by the European Union in 1993 to harmonize medical device regulations across member states.
As a Directive, MDD established general requirements that each EU member state incorporated into its own national legislation. While this improved consistency, implementation could vary slightly between countries.
Under MDD, manufacturers were required to:
For many years, MDD served as the primary regulatory framework for medical devices sold in Europe.
The Medical Device Regulation (EU) 2017/745, commonly known as MDR, was adopted in 2017 and became fully applicable on 26 May 2021.
Unlike MDD, MDR is a Regulation, meaning it applies directly and uniformly across all EU member states without requiring national implementation.
The primary objectives of MDR include:
MDR establishes a more comprehensive lifecycle approach, requiring manufacturers to monitor product performance even after CE certification has been obtained.
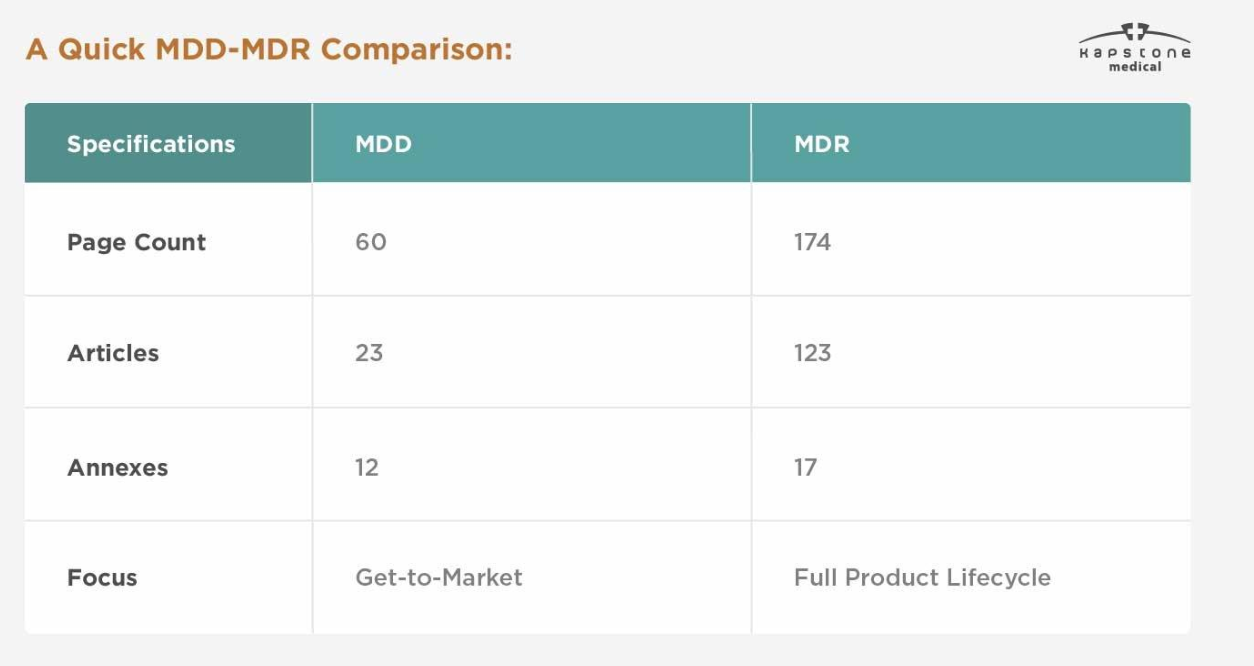
| Category | MDD | MDR |
|---|---|---|
| Legal Framework | Directive | Regulation |
| EU Reference | 93/42/EEC | (EU) 2017/745 |
| Implementation | National legislation | Directly applicable across the EU |
| Clinical Evidence | Limited requirements | Significantly enhanced requirements |
| UDI System | Not required | Mandatory for applicable devices |
| EUDAMED Registration | Not available | Required according to implementation timelines |
| Post-Market Surveillance | Basic | Comprehensive PMS & PMCF |
| Technical Documentation | Standard | More detailed and continuously maintained |
| Regulatory Oversight | Moderate | Much stricter |
The most significant change is that MDR requires manufacturers to manage compliance throughout the entire product lifecycle rather than only before market entry.
Medical technology has evolved rapidly over the past three decades. Digital dentistry, AI-assisted software, advanced biomaterials, and connected medical devices require stronger regulatory oversight than the original MDD framework was designed to provide.
MDR was introduced to:
These improvements help ensure that medical devices continue to meet safety and performance requirements after they enter the market.
The short answer is no.
New medical devices cannot obtain new MDD certificates.
Manufacturers seeking CE marking for products entering the European market must comply with MDR requirements.
Some devices that previously held valid MDD certificates have benefited from transitional arrangements established by the EU, provided specific legal conditions were met. However, these provisions are temporary and are intended to support the transition to MDR rather than replace it.
For companies planning new product launches, MDR certification is the applicable regulatory pathway.
The dental industry has experienced significant digital transformation over the last decade. As a result, many products used in modern dental laboratories fall within the scope of MDR.
Examples include:
Manufacturers must demonstrate material consistency, biocompatibility, risk management, and clinical performance through comprehensive technical documentation.
Depending on their intended medical purpose and classification, manufacturers may need to provide additional evidence relating to safety, software validation, and quality management.
Digital scanning systems often include software components that require lifecycle documentation, cybersecurity considerations, and validation processes under MDR.
Where applicable as medical devices, manufacturers must maintain complete technical documentation and implement ongoing post-market surveillance activities.
For dental manufacturers, MDR represents not only a regulatory requirement but also an opportunity to demonstrate product quality and long-term reliability.
Compared with MDD, MDR places greater emphasis on continuous compliance throughout a product's lifecycle.
Key additions include:
These requirements help improve product traceability, facilitate recalls when necessary, and provide greater confidence for healthcare professionals and patients.
Existing MDD certificates may remain valid only under specific transitional provisions established by the European Union. Manufacturers should verify whether their products qualify and monitor applicable deadlines.
No. New CE certifications for medical devices intended for the EU market must follow MDR requirements.
Yes. MDR is the primary regulatory framework governing most medical devices placed on the EU market.
Many dental materials, CAD/CAM systems, scanners, and other medical devices fall within the scope of MDR, depending on their intended purpose and classification.

MDD and MDR share the same objective—ensuring that medical devices placed on the European market are safe and effective—but they differ significantly in both structure and regulatory expectations.
While MDD established the original framework for CE certification, MDR introduces a far more comprehensive lifecycle approach, emphasizing stronger clinical evidence, improved traceability, enhanced post-market surveillance, and greater transparency.
For manufacturers of dental CAD/CAM materials and equipment, understanding MDR is no longer optional. It is an essential part of regulatory compliance, international market access, and long-term business competitiveness.
By investing in robust quality management systems, comprehensive technical documentation, and ongoing regulatory compliance, manufacturers can better meet evolving industry expectations while delivering safer, more reliable products to dental professionals worldwide.

Dry & wet milling for zirconia, PMMA, wax with auto tool changer.
learn more
High-precision 3D scanning, AI calibration, full-arch accuracy.
learn more
40-min full sintering with 57% incisal translucency and 1050 MPa strength.
learn more

40-min cycle for 60 crowns, dual-layer crucible and 200°C/min heating.
learn more
High-speed LCD printer for guides, temporaries, models with 8K resolution.
learn more







