

 中国語
中国語

デジタル総入れ歯のメリット
2024-12-31
2026-07-06
ヨーロッパで医療機器を製造、流通、購入している場合は、次のような用語に遭遇したことがあるでしょう。 MDD そして MDR。どちらも医療機器の CE マーキングに関連していますが、要件が大きく異なる 2 つの異なる規制枠組みを表しています。
以来、 医療機器規制 (EU) 2017/745 (MDR) に完全に適用されるようになりました 2021 年 5 月 26 日、徐々に前者に取って代わりました 医療機器指令 (93/42/EEC、MDD)。この移行により、臨床証拠、製品トレーサビリティ、市販後調査、技術文書に対するより厳格な要件が導入されました。
歯科用 CAD/CAM 材料および機器のメーカーにとって、コンプライアンスを維持し、欧州市場にアクセスするには、これらの違いを理解することが不可欠です。
の 医療機器指令 (93/42/EEC) 欧州連合は、加盟国全体で医療機器規制を調和させるために 1993 年に導入しました。
として 指令, MDDは、各EU加盟国が自国の国内法に組み込む一般要件を定めました。これにより一貫性が向上しましたが、実装は国によって若干異なる可能性があります。
MDD では、メーカーは次のことを要求されました。:
長年にわたり、MDD はヨーロッパで販売される医療機器の主要な規制枠組みとして機能しました。
の 医療機器規制 (EU) 2017/745一般に MDR として知られる、2017 年に採用され、完全に適用されるようになりました。 2021 年 5 月 26 日.
MDD とは異なり、MDR は 規制つまり、国内での実施を必要とせず、すべての EU 加盟国に直接かつ一律に適用されます。
MDR の主な目的は次のとおりです。:
MDR はより包括的なライフサイクル アプローチを確立し、CE 認証を取得した後でもメーカーに製品のパフォーマンスを監視することを義務付けます。
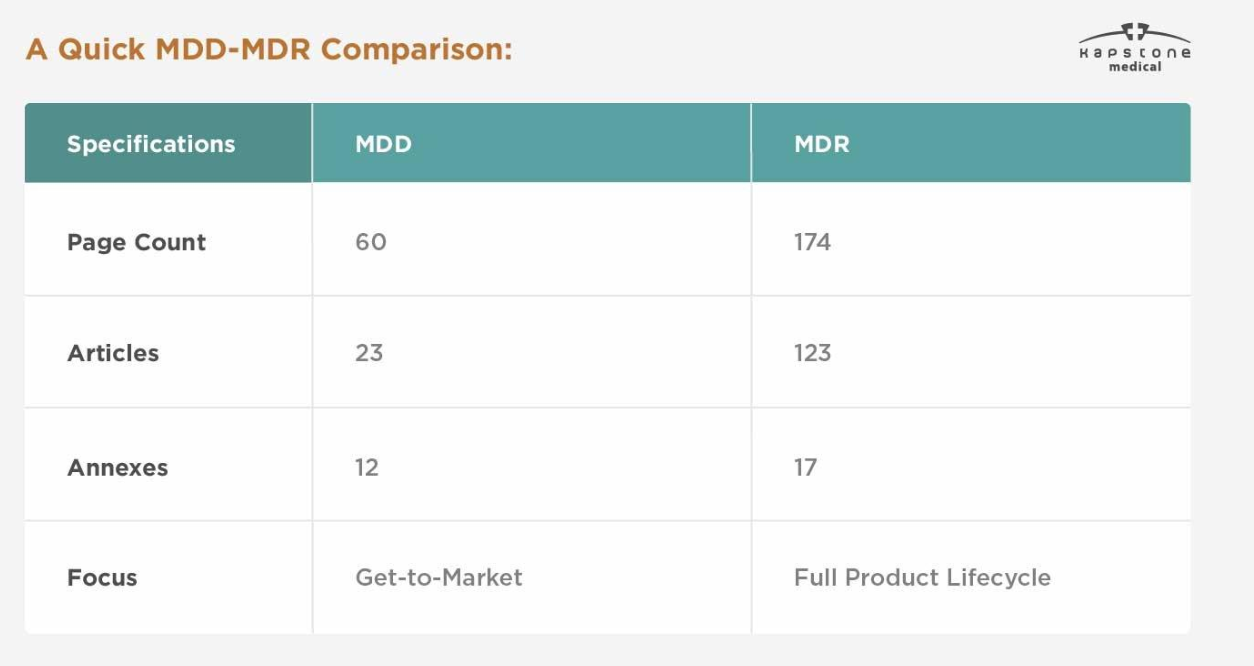
| カテゴリ | MDD | MDR |
|---|---|---|
| 法的枠組み | 指令 | 規制 |
| EU 参照 | 93/42/EEC | (EU) 2017/745 |
| 実装 | 国内法 | EU全域に直接適用可能 |
| 臨床証拠 | 限られた要件 | 大幅に強化された要件 |
| UDIシステム | 不要 | 該当するデバイスには必須 |
| EUDAMED 登録 | 利用不可 | 実装スケジュールに従って必須 |
| 市販後の監視 | 基本 | 総合的なPMSとPMCF |
| 技術文書 | 標準 | より詳細かつ継続的に保守される |
| 規制の監視 | 適度 | はるかに厳格 |
最も重要な変更は、MDR がメーカーに、市場参入前だけでなく製品ライフサイクル全体を通じてコンプライアンスを管理することを要求していることです。
医療技術は過去 30 年間で急速に進化しました。デジタル歯科、AI 支援ソフトウェア、高度な生体材料、および接続された医療機器には、当初の MDD フレームワークが提供するように設計されていたよりも強力な規制監視が必要です。
MDRが導入されました:
これらの改善により、医療機器が市場に投入された後も安全性と性能の要件を確実に満たし続けることが保証されます。
短い答えは次のとおりです いいえ.
新しい医療機器は新しい MDD 証明書を取得できません。
欧州市場に参入する製品に CE マーキングを求めるメーカーは、MDR 要件に準拠する必要があります。
以前に有効な MDD 証明書を保持していた一部のデバイスは、特定の法的条件が満たされていれば、EU によって確立された移行措置の恩恵を受けてきました。ただし、これらの規定は一時的なものであり、MDR に代わるものではなく、MDR への移行をサポートすることを目的としています。
新製品の発売を計画している企業にとって、MDR 認証は適用可能な規制経路となります。
歯科業界は過去 10 年間に大きなデジタル変革を経験しました。その結果、現代の歯科技工所で使用される多くの製品が MDR の範囲内に収まります。
例としては次のものが挙げられます。:
メーカーは、包括的な技術文書を通じて、材料の一貫性、生体適合性、リスク管理、臨床成績を証明する必要があります。
意図された医療目的と分類に応じて、メーカーは安全性、ソフトウェア検証、品質管理に関する追加の証拠を提供する必要がある場合があります。
デジタル スキャン システムには、ライフサイクルの文書化、サイバーセキュリティに関する考慮事項、MDR に基づく検証プロセスを必要とするソフトウェア コンポーネントが含まれることがよくあります。
医療機器として該当する場合、メーカーは完全な技術文書を維持し、継続的な市販後調査活動を実施する必要があります。
歯科メーカーにとって、MDR は規制要件であるだけでなく、製品の品質と長期的な信頼性を実証する機会でもあります。
MDD と比較して、MDR は製品のライフサイクル全体にわたる継続的なコンプライアンスに重点を置いています。
主な追加内容は次のとおりです。:
これらの要件は、製品のトレーサビリティを向上させ、必要な場合のリコールを容易にし、医療従事者と患者に大きな信頼を与えるのに役立ちます。
既存の MDD 証明書は、欧州連合によって確立された特定の移行規定の下でのみ有効であり続ける場合があります。メーカーは自社の製品が適格であるかどうかを確認し、適用される期限を監視する必要があります。
いいえ。EU 市場向けの医療機器の新しい CE 認証は、MDR 要件に従う必要があります。
はい。 MDR は、EU 市場に投入されるほとんどの医療機器を管理する主要な規制枠組みです。
多くの歯科材料、CAD/CAM システム、スキャナー、その他の医療機器は、その使用目的と分類に応じて MDR の範囲に含まれます。

MDD と MDR は、欧州市場に投入される医療機器の安全性と有効性を確保するという同じ目的を共有していますが、構造と規制上の期待の点で大きく異なります。
MDD は CE 認証の独自の枠組みを確立しましたが、MDR はより強力な臨床証拠、トレーサビリティの向上、市販後調査の強化、透明性の向上を強調した、より包括的なライフサイクル アプローチを導入しています。
歯科用 CAD/CAM 材料および装置のメーカーにとって、MDR を理解することはもはや必須ではありません。これは、規制遵守、国際市場へのアクセス、長期的なビジネス競争力にとって不可欠な部分です。
堅牢な品質管理システム、包括的な技術文書、継続的な規制遵守に投資することで、メーカーは進化する業界の期待にさらに応えながら、より安全で信頼性の高い製品を世界中の歯科専門家に提供することができます。














