

 中文
中文

Ventajas de la prótesis completa digital
2024-12-31
2026-07-06
Si fabrica, distribuye o compra dispositivos médicos en Europa, probablemente se haya encontrado con los términos TDM y MDR . Aunque ambos se relacionan con el marcado CE para dispositivos médicos, representan dos marcos regulatorios diferentes con requisitos significativamente diferentes.
desde el Reglamento de dispositivos médicos (UE) 2017/745 (MDR) pasó a ser plenamente aplicable en 26 mayo 2021 , ha ido sustituyendo paulatinamente al anterior Directiva sobre dispositivos médicos (93/42/CEE, MDD) . La transición ha introducido requisitos más estrictos en materia de evidencia clínica, trazabilidad de productos, vigilancia poscomercialización y documentación técnica.
Para los fabricantes de materiales y equipos CAD/CAM dentales, comprender estas diferencias es esencial para mantener el cumplimiento y acceder al mercado europeo.
El Directiva sobre dispositivos médicos (93/42/CEE) Fue introducido por la Unión Europea en 1993 para armonizar las regulaciones sobre dispositivos médicos en todos los estados miembros.
como un Directiva , MDD estableció requisitos generales que cada estado miembro de la UE incorporó a su propia legislación nacional. Si bien esto mejoró la coherencia, la implementación podría variar ligeramente entre países.
Según el MDD, los fabricantes debían:
Durante muchos años, MDD sirvió como principal marco regulatorio para los dispositivos médicos vendidos en Europa.
El Reglamento de dispositivos médicos (UE) 2017/745 , comúnmente conocido como MDR, se adoptó en 2017 y pasó a ser plenamente aplicable en 26 mayo 2021 .
A diferencia del MDD, el MDR es un Regulación , lo que significa que se aplica directa y uniformemente en todos los estados miembros de la UE sin requerir una implementación nacional.
Los objetivos principales de MDR incluyen:
MDR establece un enfoque de ciclo de vida más completo, exigiendo a los fabricantes que supervisen el rendimiento del producto incluso después de haber obtenido la certificación CE.
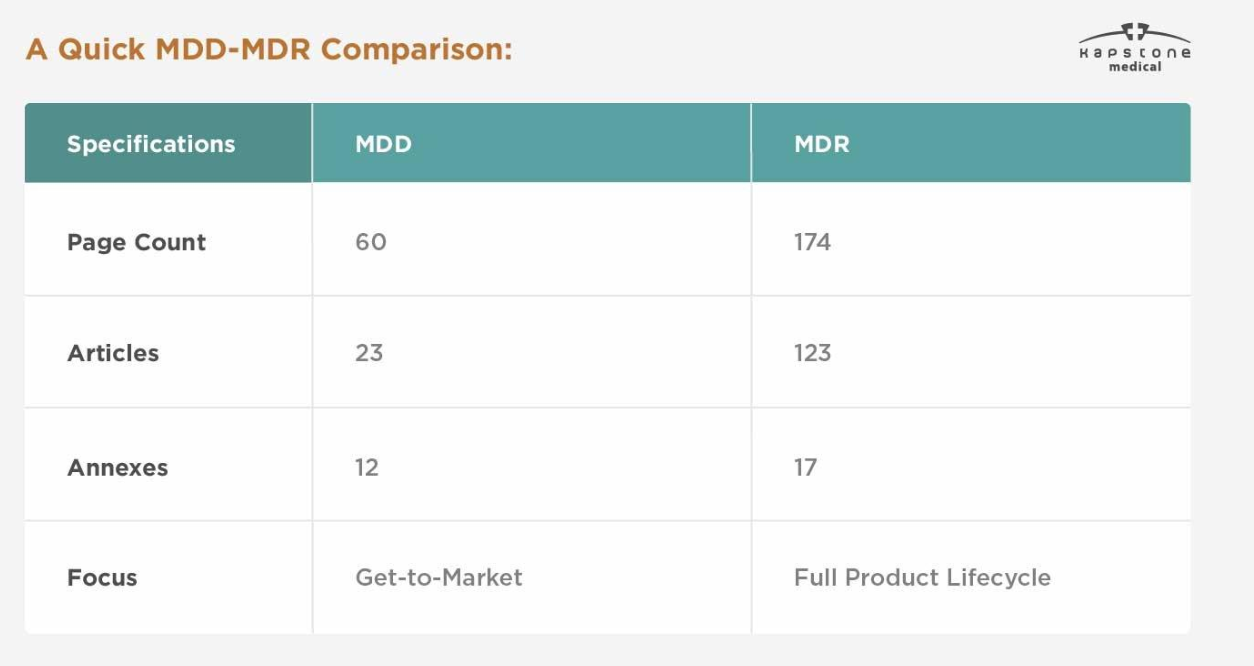
| Categoría | TDM | MDR |
|---|---|---|
| Marco Legal | Directiva | Regulación |
| Referencia UE | 93/42/EEC | (UE) 2017/745 |
| Implementación | Legislación nacional | Directamente aplicable en toda la UE |
| Evidencia clínica | Requisitos limitados | Requisitos significativamente mejorados |
| Sistema UDI | No requerido | Obligatorio para los dispositivos aplicables |
| Registro EUDAMED | No disponible | Requerido según los cronogramas de implementación |
| Vigilancia posterior a la comercialización | Básico | PMS y PMCF integrales |
| Documentación técnica | Estándar | Más detallado y mantenido continuamente |
| Supervisión regulatoria | Moderado | Mucho más estricto |
El cambio más significativo es que MDR requiere que los fabricantes gestionen el cumplimiento durante todo el ciclo de vida del producto y no solo antes de su entrada al mercado.
La tecnología médica ha evolucionado rápidamente durante las últimas tres décadas. La odontología digital, el software asistido por IA, los biomateriales avanzados y los dispositivos médicos conectados requieren una supervisión regulatoria más estricta que la que el marco MDD original fue diseñado para proporcionar.
MDR fue introducido a:
Estas mejoras ayudan a garantizar que los dispositivos médicos sigan cumpliendo con los requisitos de seguridad y rendimiento después de su ingreso al mercado.
La respuesta corta es No .
Los dispositivos médicos nuevos no pueden obtener nuevos certificados MDD.
Los fabricantes que buscan el marcado CE para productos que ingresan al mercado europeo deben cumplir con los requisitos del MDR.
Algunos dispositivos que anteriormente tenían certificados MDD válidos se han beneficiado de acuerdos transitorios establecidos por la UE, siempre que se cumplieran condiciones legales específicas. Sin embargo, estas disposiciones son temporales y están destinadas a apoyar la transición al MDR en lugar de reemplazarlo.
Para las empresas que planean el lanzamiento de nuevos productos, la certificación MDR es la vía regulatoria aplicable.
La industria dental ha experimentado una importante transformación digital durante la última década. Por ello, muchos productos utilizados en los laboratorios dentales modernos entran dentro del ámbito de aplicación del MDR.
Los ejemplos incluyen:
Los fabricantes deben demostrar la consistencia del material, la biocompatibilidad, la gestión de riesgos y el rendimiento clínico a través de documentación técnica completa.
Dependiendo de su clasificación y propósito médico previsto, es posible que los fabricantes deban proporcionar evidencia adicional relacionada con la seguridad, la validación del software y la gestión de calidad.
Los sistemas de escaneo digital a menudo incluyen componentes de software que requieren documentación del ciclo de vida, consideraciones de ciberseguridad y procesos de validación según MDR.
Cuando corresponda como dispositivos médicos, los fabricantes deben mantener la documentación técnica completa e implementar actividades continuas de vigilancia posterior a la comercialización.
Para los fabricantes dentales, MDR representa no sólo un requisito reglamentario sino también una oportunidad para demostrar la calidad del producto y la confiabilidad a largo plazo.
En comparación con MDD, MDR pone mayor énfasis en el cumplimiento continuo durante todo el ciclo de vida de un producto.
Las adiciones clave incluyen:
Estos requisitos ayudan a mejorar la trazabilidad del producto, facilitan las retiradas cuando sea necesario y proporcionan mayor confianza a los profesionales sanitarios y a los pacientes.
Los certificados MDD existentes pueden seguir siendo válidos sólo bajo disposiciones transitorias específicas establecidas por la Unión Europea. Los fabricantes deben verificar si sus productos califican y monitorear los plazos aplicables.
No. Las nuevas certificaciones CE para dispositivos médicos destinados al mercado de la UE deben seguir los requisitos del MDR.
Sí. MDR es el principal marco regulatorio que rige la mayoría de los dispositivos médicos comercializados en el mercado de la UE.
Muchos materiales dentales, sistemas CAD/CAM, escáneres y otros dispositivos médicos entran dentro del alcance de MDR, dependiendo de su propósito y clasificación.

MDD y MDR comparten el mismo objetivo (garantizar que los dispositivos médicos comercializados en el mercado europeo sean seguros y eficaces), pero difieren significativamente tanto en estructura como en expectativas regulatorias.
Si bien MDD estableció el marco original para la certificación CE, MDR introduce un enfoque de ciclo de vida mucho más integral, enfatizando evidencia clínica más sólida, trazabilidad mejorada, vigilancia posterior a la comercialización mejorada y mayor transparencia.
Para los fabricantes de materiales y equipos CAD/CAM dentales, comprender MDR ya no es opcional. Es una parte esencial del cumplimiento normativo, el acceso a los mercados internacionales y la competitividad empresarial a largo plazo.
Al invertir en sistemas sólidos de gestión de calidad, documentación técnica integral y cumplimiento normativo continuo, los fabricantes pueden satisfacer mejor las expectativas cambiantes de la industria y, al mismo tiempo, ofrecer productos más seguros y confiables a los profesionales dentales de todo el mundo.

Fresado en seco y húmedo para circonio, PMMA y cera con cambiador automático de herramientas.
aprender más
Escaneo 3D de alta precisión, calibración AI, precisión de arco completo.
aprender más
Sinterización completa en 40 minutos con 57% de translucidez incisal y resistencia de 1050 MPa.
aprender más
Escáner ultrarrápido de precisión de 5 micrones con exportación STL abierta.
aprender más
Ciclo de 40 min para 60 coronas, crisol bicapa y calentamiento a 200°C/min.
aprender más
Impresora LCD de alta velocidad para guías, temporales, modelos con resolución 8K.
aprender más







