

 chinesisch
chinesisch

Vorteile der digitalen Totalprothese
2024-12-31
2026-07-06
Wenn Sie in Europa medizinische Geräte herstellen, vertreiben oder kaufen, sind Ihnen die Begriffe wahrscheinlich schon begegnet MDD Und MDR . Obwohl sich beide auf die CE-Kennzeichnung von Medizinprodukten beziehen, stellen sie zwei unterschiedliche Regulierungsrahmen mit deutlich unterschiedlichen Anforderungen dar.
Seit dem Medizinprodukteverordnung (EU) 2017/745 (MDR) wurde am voll anwendbar 26. Mai 2021 , es hat nach und nach das erstere ersetzt Medizinprodukterichtlinie (93/42/EWG, MDD) . Durch den Übergang wurden strengere Anforderungen an klinische Beweise, Produktrückverfolgbarkeit, Überwachung nach dem Inverkehrbringen und technische Dokumentation eingeführt.
Für Hersteller von zahnmedizinischen CAD/CAM-Materialien und -Geräten ist das Verständnis dieser Unterschiede für die Einhaltung der Vorschriften und den Zugang zum europäischen Markt von entscheidender Bedeutung.
Der Medizinprodukterichtlinie (93/42/EWG) wurde 1993 von der Europäischen Union eingeführt, um die Vorschriften für Medizinprodukte in den Mitgliedstaaten zu harmonisieren.
Als Richtlinie Mit der MDD wurden allgemeine Anforderungen festgelegt, die jeder EU-Mitgliedstaat in seine eigene nationale Gesetzgebung umsetzte. Obwohl dadurch die Konsistenz verbessert wurde, kann die Umsetzung von Land zu Land leicht variieren.
Im Rahmen der MDD waren die Hersteller dazu verpflichtet:
Viele Jahre lang diente MDD als primärer Regulierungsrahmen für in Europa verkaufte Medizinprodukte.
Der Medizinprodukteverordnung (EU) 2017/745 , allgemein bekannt als MDR, wurde 2017 verabschiedet und ist ab dem 1. April 2017 in vollem Umfang anwendbar 26. Mai 2021 .
Im Gegensatz zu MDD ist MDR ein Verordnung Dies bedeutet, dass es in allen EU-Mitgliedstaaten unmittelbar und einheitlich gilt, ohne dass eine nationale Umsetzung erforderlich ist.
Zu den Hauptzielen der MDR gehören::
MDR etabliert einen umfassenderen Lebenszyklusansatz und verlangt von Herstellern, die Produktleistung auch nach Erhalt der CE-Zertifizierung zu überwachen.
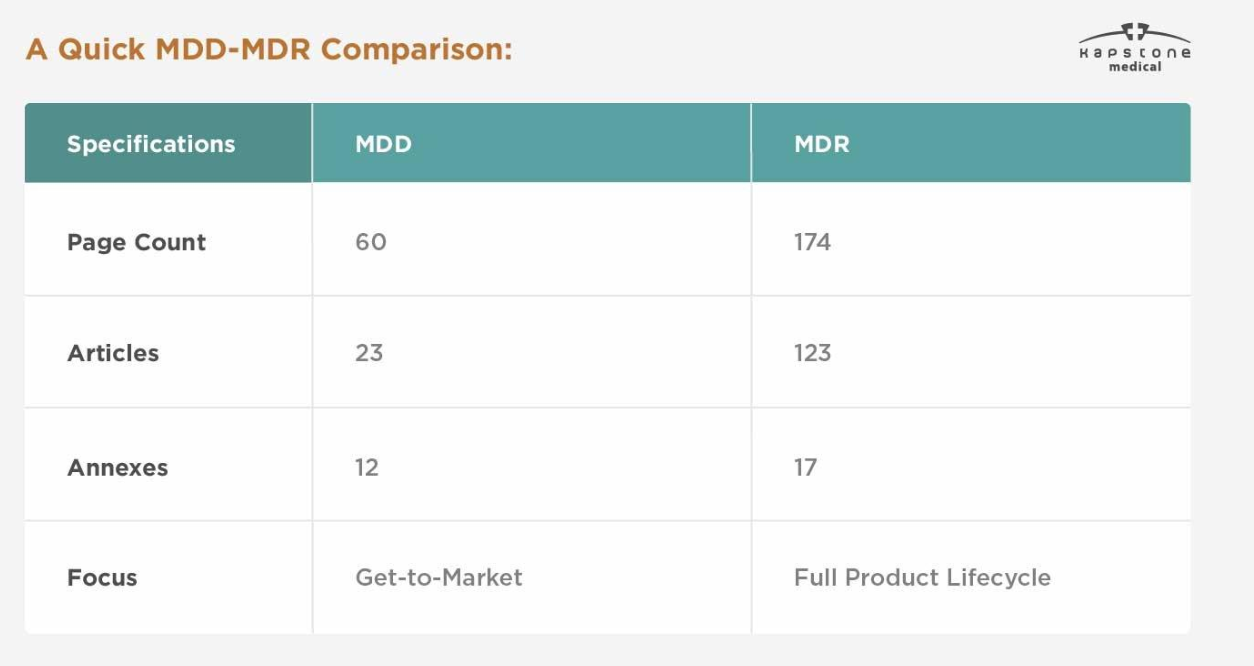
| Kategorie | MDD | MDR |
|---|---|---|
| Rechtlicher Rahmen | Richtlinie | Verordnung |
| EU-Referenz | 93/42/EEC | (EU) 2017/745 |
| Durchführung | Nationale Gesetzgebung | Direkt anwendbar in der gesamten EU |
| Klinischer Beweis | Begrenzte Anforderungen | Deutlich erhöhte Anforderungen |
| UDI-System | Nicht erforderlich | Obligatorisch für entsprechende Geräte |
| EUDAMED-Registrierung | Nicht verfügbar | Erforderlich gemäß den Implementierungsfristen |
| Überwachung nach dem Inverkehrbringen | Basic | Umfassendes PMS und PMCF |
| Technische Dokumentation | Standard | Detaillierter und kontinuierlich gepflegt |
| Regulierungsaufsicht | Mäßig | Viel strenger |
Die bedeutendste Änderung besteht darin, dass die MDR von den Herstellern verlangt, die Einhaltung der Vorschriften während des gesamten Produktlebenszyklus und nicht nur vor dem Markteintritt zu verwalten.
Die Medizintechnik hat sich in den letzten drei Jahrzehnten rasant weiterentwickelt. Digitale Zahnheilkunde, KI-gestützte Software, fortschrittliche Biomaterialien und vernetzte medizinische Geräte erfordern eine strengere regulatorische Aufsicht, als sie im ursprünglichen MDD-Rahmen vorgesehen war.
MDR wurde eingeführt:
Diese Verbesserungen tragen dazu bei, dass medizinische Geräte auch nach ihrer Markteinführung weiterhin den Sicherheits- und Leistungsanforderungen entsprechen.
Die kurze Antwort lautet NEIN .
Neue Medizinprodukte können keine neuen MDD-Zertifikate erhalten.
Hersteller, die eine CE-Kennzeichnung für Produkte anstreben, die auf den europäischen Markt kommen, müssen die MDR-Anforderungen erfüllen.
Einige Geräte, die zuvor über gültige MDD-Zertifikate verfügten, profitierten von Übergangsregelungen der EU, sofern bestimmte rechtliche Bedingungen erfüllt waren. Diese Bestimmungen sind jedoch vorübergehender Natur und sollen den Übergang zur MDR unterstützen und nicht ersetzen.
Für Unternehmen, die die Einführung neuer Produkte planen, ist die MDR-Zertifizierung der anwendbare regulatorische Weg.
Die Dentalbranche hat im letzten Jahrzehnt einen erheblichen digitalen Wandel erlebt. Daher fallen viele Produkte, die in modernen Dentallaboren zum Einsatz kommen, in den Geltungsbereich der MDR.
Beispiele hierfür sind:
Hersteller müssen Materialkonsistenz, Biokompatibilität, Risikomanagement und klinische Leistung durch umfassende technische Dokumentation nachweisen.
Abhängig von ihrem beabsichtigten medizinischen Zweck und ihrer Klassifizierung müssen Hersteller möglicherweise zusätzliche Nachweise in Bezug auf Sicherheit, Softwarevalidierung und Qualitätsmanagement erbringen.
Digitale Scansysteme umfassen häufig Softwarekomponenten, die eine Lebenszyklusdokumentation, Überlegungen zur Cybersicherheit und Validierungsprozesse gemäß MDR erfordern.
Sofern es sich um Medizinprodukte handelt, müssen Hersteller eine vollständige technische Dokumentation führen und fortlaufende Überwachungsaktivitäten nach dem Inverkehrbringen durchführen.
Für Dentalhersteller stellt MDR nicht nur eine regulatorische Anforderung dar, sondern auch eine Gelegenheit, Produktqualität und langfristige Zuverlässigkeit unter Beweis zu stellen.
Im Vergleich zur MDD legt die MDR größeren Wert auf die kontinuierliche Einhaltung der Vorschriften über den gesamten Lebenszyklus eines Produkts.
Zu den wichtigsten Ergänzungen gehören::
Diese Anforderungen tragen dazu bei, die Rückverfolgbarkeit von Produkten zu verbessern, Rückrufe bei Bedarf zu erleichtern und das Vertrauen von medizinischem Fachpersonal und Patienten zu stärken.
Bestehende MDD-Zertifikate können nur unter bestimmten, von der Europäischen Union festgelegten Übergangsbestimmungen gültig bleiben. Hersteller sollten überprüfen, ob ihre Produkte die Anforderungen erfüllen, und die geltenden Fristen überwachen.
Nein. Neue CE-Zertifizierungen für Medizinprodukte, die für den EU-Markt bestimmt sind, müssen den MDR-Anforderungen entsprechen.
Ja. Die MDR ist der primäre Regulierungsrahmen für die meisten Medizinprodukte, die in der EU auf den Markt gebracht werden.
Viele Dentalmaterialien, CAD/CAM-Systeme, Scanner und andere medizinische Geräte fallen je nach Verwendungszweck und Klassifizierung in den Geltungsbereich der MDR.

MDD und MDR haben das gleiche Ziel – sicherzustellen, dass auf den europäischen Markt gebrachte Medizinprodukte sicher und wirksam sind –, unterscheiden sich jedoch sowohl in der Struktur als auch in den regulatorischen Erwartungen erheblich.
Während MDD den ursprünglichen Rahmen für die CE-Zertifizierung festlegte, führt MDR einen weitaus umfassenderen Lebenszyklusansatz ein, der stärkere klinische Beweise, verbesserte Rückverfolgbarkeit, verbesserte Überwachung nach dem Inverkehrbringen und größere Transparenz hervorhebt.
Für Hersteller von zahnmedizinischen CAD/CAM-Materialien und -Geräten ist das Verständnis von MDR nicht mehr optional. Es ist ein wesentlicher Bestandteil der Einhaltung gesetzlicher Vorschriften, des internationalen Marktzugangs und der langfristigen Wettbewerbsfähigkeit von Unternehmen.
Durch die Investition in robuste Qualitätsmanagementsysteme, eine umfassende technische Dokumentation und die kontinuierliche Einhaltung gesetzlicher Vorschriften können Hersteller die sich ändernden Erwartungen der Branche besser erfüllen und gleichzeitig sicherere und zuverlässigere Produkte für Zahnärzte auf der ganzen Welt liefern.

Trocken- und Nassfräsen für Zirkonoxid, PMMA, Wachs mit automatischem Werkzeugwechsler.
Erfahren Sie mehr
Hochpräzises 3D-Scannen, KI-Kalibrierung, Genauigkeit des gesamten Zahnbogens.
Erfahren Sie mehr
40-minütiges vollständiges Sintern mit 57 % inzisaler Transluzenz und 1050 MPa Festigkeit.
Erfahren Sie mehr
Ultraschneller 5-Mikron-Genauigkeitsscanner mit offenem STL-Export.
Erfahren Sie mehr
40-Minuten-Zyklus für 60 Kronen, zweischichtiger Tiegel und 200 °C/Minute Erhitzen.
Erfahren Sie mehr
Hochgeschwindigkeits-LCD-Drucker für Anleitungen, Provisorien und Modelle mit 8K-Auflösung.
Erfahren Sie mehr







